Novel FoldDock pipeline uses AlphaFold2 to provide accurate predictions of heterodimeric complexes structures
Reliably determining the three-dimensional (3D) structure of proteins has long since been considered one of the major challenges of modern-day biology. Understanding the 3D structure of proteins is believed to be key in, for example, elucidating previously unknown protein functions and gaining insight into protein-protein interactions. The ability to reliably predict 3D protein structure is therefore crucial in deepening our understanding of many fields, including human biology, drug discovery, antibiotic resistance, environmental challenges, and climate change.
Until recently, all computational methods used to predict 3D protein structure have been based either on physical interactions or evolutionary history. The former method is challenged in multiple ways, including the computational intractability of molecular simulation, and the difficulty involved in producing accurate models of protein physics. The latter method has primarily involved a bioinformatics approach; using evolutionary relatedness or homology to proteins with known structures to make inferences about proteins of unknown structure. This was made possible, in part, due to the large number of protein structures deposited in the Protein Data Bank (PDB). Neither method has reliably predicted 3D protein structure though, and so their use has been limited.
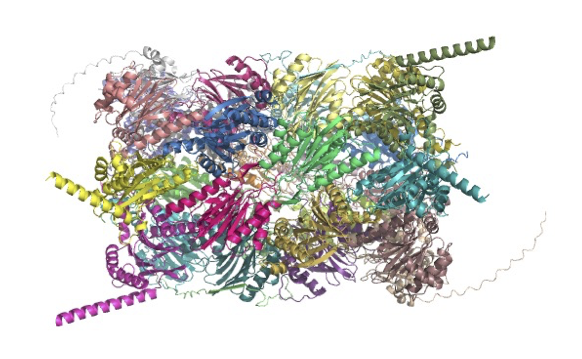
In recent years, AlphaFold, an AI system developed by DeepMind, has revolutionized computational protein structure prediction in many ways. AlphaFold allows for greater, near experimental, speed and accuracy compared to earlier predictive methods. In addition, DeepMind in partnership with EMBL’s European Bioinformatics Institute (EMBL-EBI), have made the resultant predictions openly available to researchers worldwide. Alphafold is able to predict single-chain protein structure with a high degree of accuracy. The main limitation of Alphafold is that it cannot accurately predict the structures of protein complexes. This is partly related to the fact that, although protein-protein interactions are central to biology, their complexity and the difficultly involved in performing docking has hampered both experimental and computational research efforts.
In a recent article researchers from SciLifeLab and the Department of Biochemistry and Biophysics at Stockholm University (first authors: Patrick Bryant and Gabriele Pozzati; corresponding author: Arne Elofsson) improved the prediction of protein-protein interactions using AlphaFold2. AlphaFold2 is team name of the neural network AlphaFold entered into the CASP14 assessment (May-July 2020).
In their previous studies (Pozatti et al. 2021; Lamb & Elofsson, 2021) the Elofsson research group has used distance and angle constraints from trRosetta to predict the interaction between two chains from a heterodimeric protein complex. Whilst knowledge about intra-chain structural feature may be successfully extended to derive inter-chain features, less than one in ten tested proteins were found to fold and dock in the correct way. However, multiple sequence alignment (MSA) was found to be a crucial part of the correct Dock and fold approach.
In their latest study, the researchers decided to apply the AlphaFold2 (AF2) pipeline on two different datasets to Fold and Dock protein-protein pairs at the same time. In addition, the researchers tested docking with the AF2 pipeline in combination with different input MSAs to evaluate whether there is a correlation between the output model quality and MSA inputs. Bryant et al. showed that by combining the AF2 protocol with optimized MSAs, and repeating scoring of models of the same protein-protein interaction with a predicted DockQ score, they were able to generate protein-protein interaction models with acceptable quality (DockQ ≥ 0.23) for 63% of the dimers. In addition, the predicted interfaces were used to form a function to predict the DockQ score and determine whether a model is acceptable or incorrect. Using the predicted DockQ scores, the researchers were able to identify half of the interacting pairs at 1% FPR.
In summary, Bryant and colleagues showed that, by using an optimized MSAs with AF2, they could perform accurate predictions of the structures of heterodimeric complexes with an SR (e.g., percentage of acceptable models) of 62.7% on a large test set). The MSAs were found to be a crucial part of the pipeline, but may also be the limiting factor. However by using fast MSA generation this can be solved. The novel pipeline outlined in this study, called FoldDock, has the potential for rapidly expanding our understanding of known structural protein interactions at a low cost.
Dataset
- The raw data used in this study, including MSAs and predicted PDB files, can be found in SciLifeLab Data Repository under DOI: 10.17044/scilifelab.16866202.
- All code to run FoldDock and reproduce the analysis here can be obtained here (commit 2e4c96aa352338976260ece0646ceaaa75392dec) under the Apache License, Version 2.0.
Article
Bryant, P., Pozzati, G. Elofsson, A. (2022). Improved prediction of protein-protein interactions using AlphaFold2. Nature Communications, 13, 1265. 10.1038/s41467-022-28865-w
Funding
The research project was funded by Swedish Research Council for Natural Science, Swedish E-science Research Center. The Computational resources used were from Swedish National Infrastructure for Computing, SNIC and Berzelius.