aMeta; a new metagenomic profiling workflow for ancient DNA
The development of ancient DNA (aDNA) technologies has contributed to new insights into, for example, human and faunal evolution and demography. Multiple important discoveries about ancient populations have recently been made using next-generation sequencing (NGS) technologies. For example, it has been shown that host-associated microbial aDNA from eukaryotic remains can provide important information about ancient lifestyles, how populations migrated, and ancient diseases and pandemics. The analysis of microbial aDNA from different archaeological samples is now a growing research field, as is the study of sedimentary ancient DNA (sedaDNA). SedaDNA has quickly become an independent field within palaeogenetics, and allows studies on, for example, how marine ecosystems developed over time. SedaDNA offers unprecedented discoveries about the evolution of hominins and animals, without the need to analyse historical bones or teeth.
Ancient metagenomics has contributed to many new findings. However, the accurate detection, abundance quantification, and authentication analysis of microbial organisms in ancient metagenomic samples remains challenging. The main issue in aDNA metagenomics studies is that the amount of microbial aDNA from a specific sample is limited. Further, the samples are often contaminated by other host or invasive microbial communities, or due to technical factors in modern times. Identifying ancient microbes is therefore difficult, and most ancient metagenomic studies have a high rate of false-positive and false-negative microbial identifications. One way to solve the issue with high error rates is by using more stringent computational frameworks, but few ancient metagenomic computational workflows have been available before now.
In a recent publication in Genome Biology, researchers from SciLifeLab, NBIS, Stockholm University, Centre for Palaeogenetics, Swedish Museum of Natural History, Linköping University, Lund University, and international collaborators (First authors: Zoé Pochon, Nora Bergfeldt; Last authors: Anders Götherström, Claudio Mirabello, Per Unneberg, and Nikolay Oskolkov; Corresponding author: Nikolay Oskolkov) proposed and benchmarked a new metagenomic workflow for aDNA.
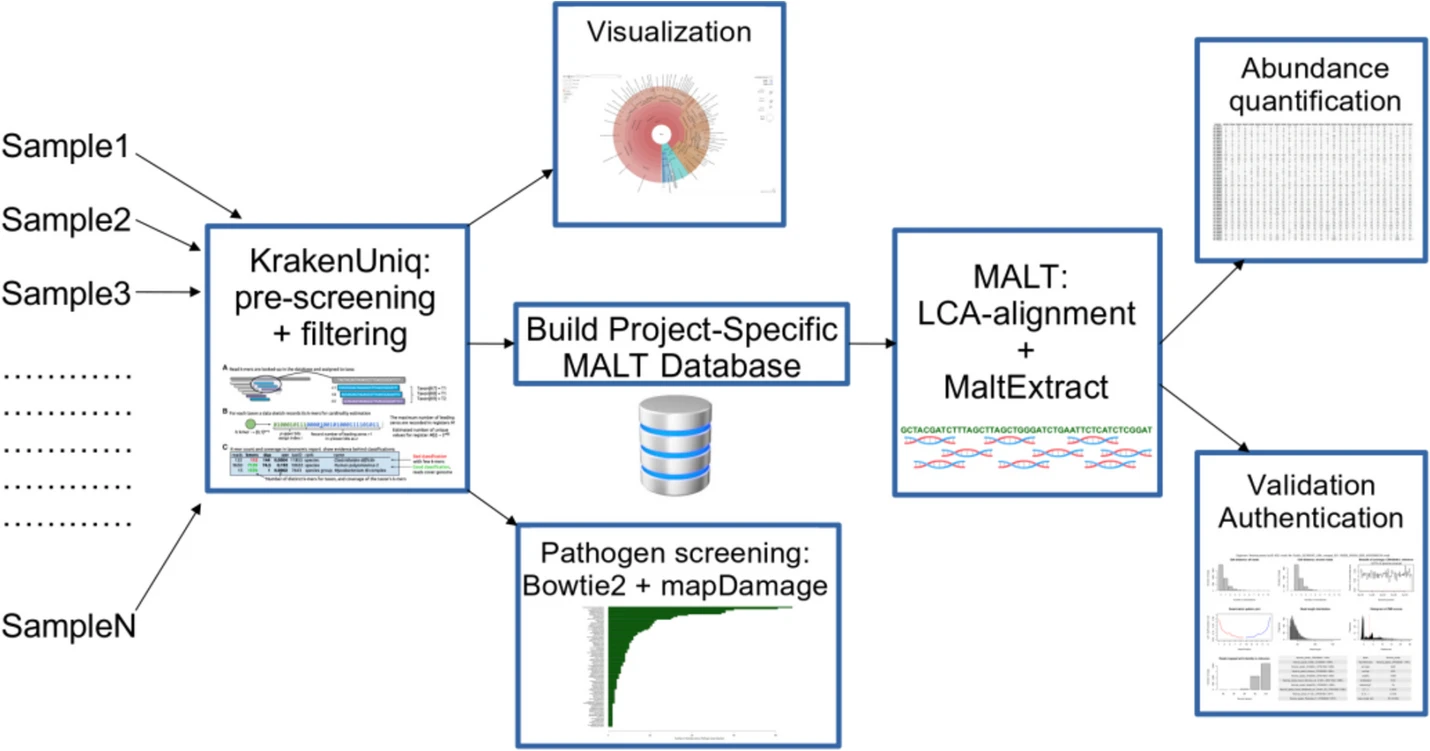
Pochon and colleagues developed a metagenomic workflow called aMeta, designed to both reduce the risk of false discoveries, and minimise computer memory requirements. In ancient metagenomics, two types of error occur regularly: detection errors (it is hard to correctly identify microbial presence in a metagenomic sample), and authentication errors (common in ancient samples and may be caused by modern contamination during processing and handling, which can lead to a modern microbe being studied). The aim of the aMeta workflow is therefore to establish whether an ancient microbe is present in the sample.
Pochon et al. combined both classification- and alignment-based approaches in the aMeta workflow. They used KrakenUniq, compatible with low-memory computer use, and a Least Common Ancestor (LCA)-based MALT alignment. The workflow was written using the Snakemake language for workflow management to ensure reproducibility, and the final workflow documentation and code were shared openly on GitHub and Zenodo. The results showed that a KrakenUniq-based selection of microbial candidates for inclusion in the MALT database reduced the resource usage of aMeta when compared to using metagenomic profiling with MALT.
The researchers benchmarked aMeta against Heuristic Operations for Pathogen Screening (HOPS); a common, popular de facto standard for ancient metagenomics. The researchers showed that aMeta had a higher sensitivity vs. specificity balance for the detection and authentication of ancient microbes when compared to HOPS using the same amount of computer memory. This was possible as KrakenUniq and MALT had a greater database size and flexible authentication scoring system. Notably, aMeta consumed about half as much computer memory compared to HOPS when tested with a benchmark simulated ancient metagenomic dataset.
“The peculiarity of ancient microbiome analysis is that unbiased detection of ancient microbes requires large reference databases which take many terabytes of computer memory, which is typically not available in many labs”, says corresponding author Nikolay Oskolkov from NBIS. “Therefore aMeta attempts to optimise resource usage whilst still operating with large databases, i.e. without compromising the accuracy of metagenomic analysis.”.
In summary, Pochon et al. developed a new metagenomic profiling workflow for ancient DNA, called aMeta. This workflow was benchmarked against the de facto standard, HOPS, which is the current state-of-the-art workflow. The new aMeta workflow was found to be superior in microbial detection and authentication, and to require a lower usage of computer memory.
Article
DOI: https://doi.org/10.1186/s13059-023-03083-9
Pochon, Z., Bergfeldt, N., Kırdök, E., Vicente, M., Naidoo, T., van der Valk, T., Altınışık, N. E., Krzewińska, M., Dalén, L., Götherström, A., Mirabello, C., Unneberg, P., Oskolkov, N. (2023) aMeta: an accurate and memory-efficient ancient metagenomic profiling workflow. In: Genome Biology (Vol: 24, Article no.: 242).
Funders
Funding was provided by the Swedish Research Council, and Knut and Alice Wallenberg Foundation as part of the National Bioinformatics Infrastructure Sweden at SciLifeLab.
Data and code availability
- The workflow is publicly available on GitHub and deposited in a Zenodo repository.
- The non-redundant NCBI NT KrakenUniq database can be accessed on the SciLifeLab Data Repository.
- The microbial version of NCBI NT combined with human and complete eukaryotic reference genomes can be accessed on the SciLifeLab Data Repository.
Infrastructure
The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC), partially funded by the Swedish Research Council.