New pipeline for mRNA-Seq and ATAC-Seq analysis allows for biological insights without in-depth bioinformatics skills
The costs for next-generation sequencing (NGS) have decreased in recent years, making omics-scale assays more affordable. The development of efficient and reproducible analysis pipelines (e.g. Nextflow pipelines), and package managers (e.g. Docker, Singularity etc.) have also contributed to making NGS methods more accessible for the research community.
Two common methods, mRNA-Seq (which measures gene expression), and ATAC-Seq (which measures chromatin accessibility) are often used for omics research. Today, a number of pipelines are available to analyse either mRNA-Seq and ATAC-Seq data. However, when assessing the main features and implementation of published ATAC-Seq analysis pipeline tools, it is clear that most have limited options for downstream analyses, such as enrichment analysis and integration with mRNA-Seq data. To perform such analyses, researchers therefore require advanced bioinformatics skills.
In the July 2024 edition of Genomics, researchers from Karolinska Institutet and National Genomics Infrastructure (NGI) Sweden (a SciLifeLab infrastruture unit) proposed a new pipeline that can be used for comprehensive ATAC-Seq and mRNA-Seq data analysis (Salignon et al., 2024). This analysis pipeline is named Cactus (Chromatin accessibility and transcriptomics unifying software). It allows researchers to perform efficient and reproducible analyses of interrelated changes in chromatin accessibility and gene expression, without the need for advanced bioinformatics skills.
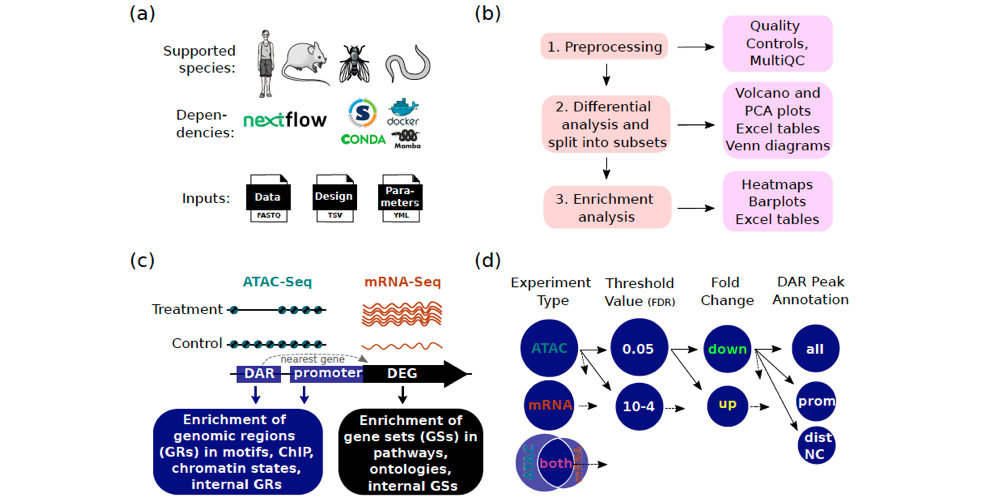
Cactus is coded in Nextflow, and all tools are encapsulated within containers (e.g. Docker or Singularity) or virtual environments (e.g. conda or Mamba). This architecture allows for efficient and reproducible analyses. The Cactus pipeline can analyse mRNA-Seq or ATAC-Seq data either separately or jointly. The pipeline initially acts to preprocess the raw reads. It then performs differential analyses (DA), and divides the results into subsets (Differential Analysis Subsets (DASs)). Lastly, the pipeline performs enrichment analysis on each subset, which allows for comprehensive molecular insights.
The researchers compared the Cactus pipeline with other published ATAC-Seq analysis tools and found that, although some features are available in other tools, Cactus provides multiple unique functionalities. This includes, for example, splitting the DA results into subsets, and enrichment in chromatin states and ChIP-Seq binding sites. Cactus also allows for the creation of customised heatmaps, as well as the reporting of results in merged pdfs and tables.
Salignon et al. (2024) used Cactus to analyse a previously published dataset containing samples from Caenorhabditis elegans and human cells. Cactus confirmed the main findings of the previously published research, and provided additional observations not found in the original study.
“Cactus stands out amongst other ATAC-Seq pipelines in its multi-modal, visualisation, and downstream analysis abilities. Furthermore, it aims to be as simple to use as possible, with one-liner calls, while being highly customisable through various parameters. We hope that these features will enable researchers, especially from labs with limited bioinformatic expertise, to quickly explore their data through various angles.” says corresponding author Jérôme Salignon from Karolinska Institutet.
Researchers in Sweden can easily run Cactus on various NAISS resources by loading the cactus_atac module, which is installed on Rackham, Bianca, Miarka, and Snowy. An example on how to do this is provided on the Cactus documentation.
In summary, Cactus is an analysis pipeline that can assist researchers in gaining comprehensive insights from chromatin accessibility and gene expression data. It has multiple unique features compared to other ATAC-Seq data analysis tools. The researchers have shared the code and software for Cactus openly, making this a useful tool for omics data analysis.
Article
DOI: 10.1016/j.ygeno.2024.110858
Salignon, J., Millan-Ariño, L., Garcia, M. U., & Riedel C. G. (2024) Cactus: A user-friendly and reproducible ATAC-Seq and mRNA-Seq analysis pipeline for data preprocessing, differential analysis, and enrichment analysis. In: Genomics (Vol: 116, Issue: 4, 110858).
Funders
Funding was provided by Swedish Research Council (VR) grants, a Swedish Cancer Society (Cancerfonden) grant, and Novo Nordisk Foundation grants.
Data and code availability
- The code and documentation for the Cactus workflow are available on Github, as well as in the Zenodo open repository as a release archive.
- The published dataset containing samples from Caenorhabditis elegans and human cells, which was used to validate the Cactus pipeline, is held in the Gene Expression Omnibus (GEO) database, and is available under the GEO accession GSE98758.
- Test datasets and references are available as a release archive in the SciLifeLab Data Repository under DOI: 10.17044/scilifelab.20171347.v4.
Infrastructure
The SciLifeLab team at National Genomics Infrastructure (NGI) Sweden contributed to this study.